
Computational Biophysics & The Disordered Proteome

Michael Bakker, B.Sc., M.Sc., Ph.D.
Post-Doctoral Researcher in Biophysics and Physical Chemistry
Hello. I’m a computational scientist working at the intersection of structural biology and machine learning to investigate the molecular mechanisms underlying human disease. My research uses dimensionality reduction and clustering to create more accurate structural ensembles for proteins. Key biological areas of my work include predicting the behavior of DNA Binding Domains, analyzing Polyproline Rich Regions, and computationally targeting orphan nuclear receptors.
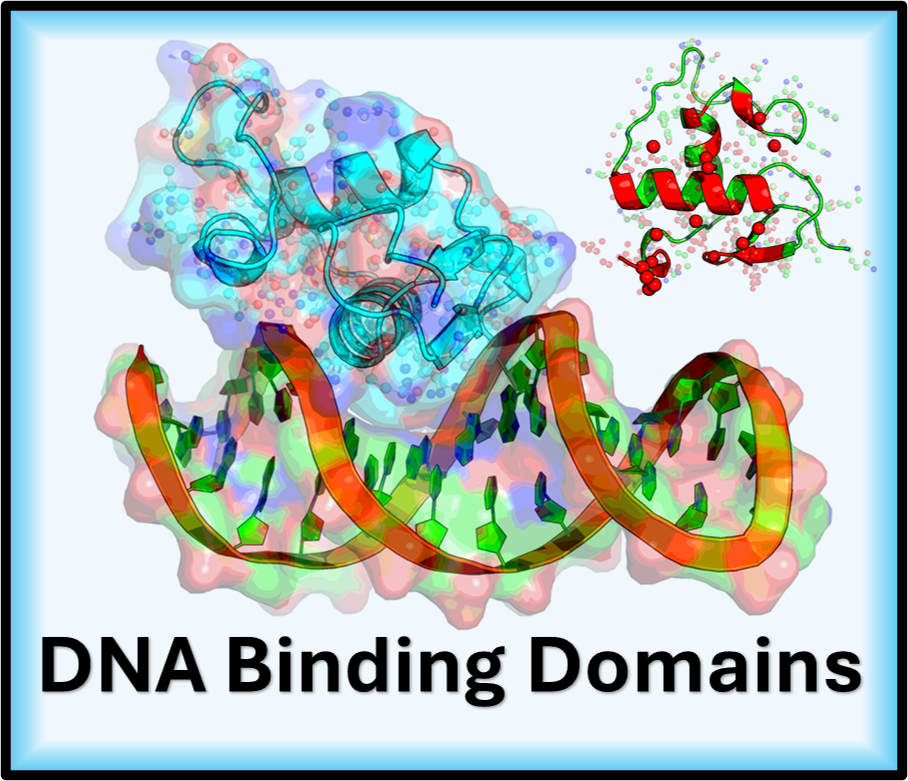
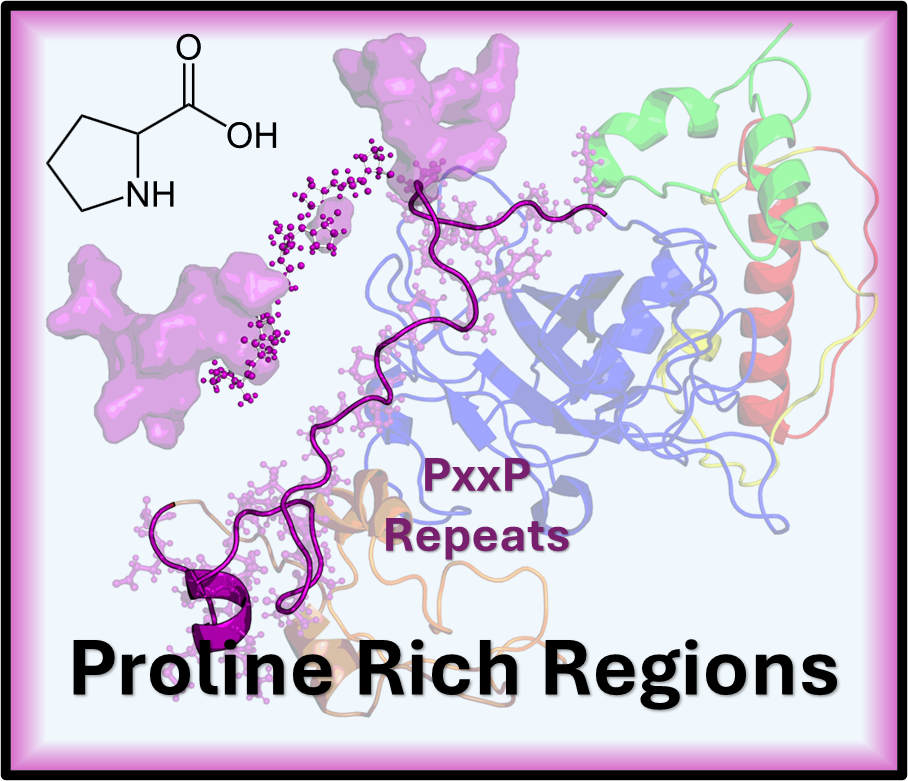
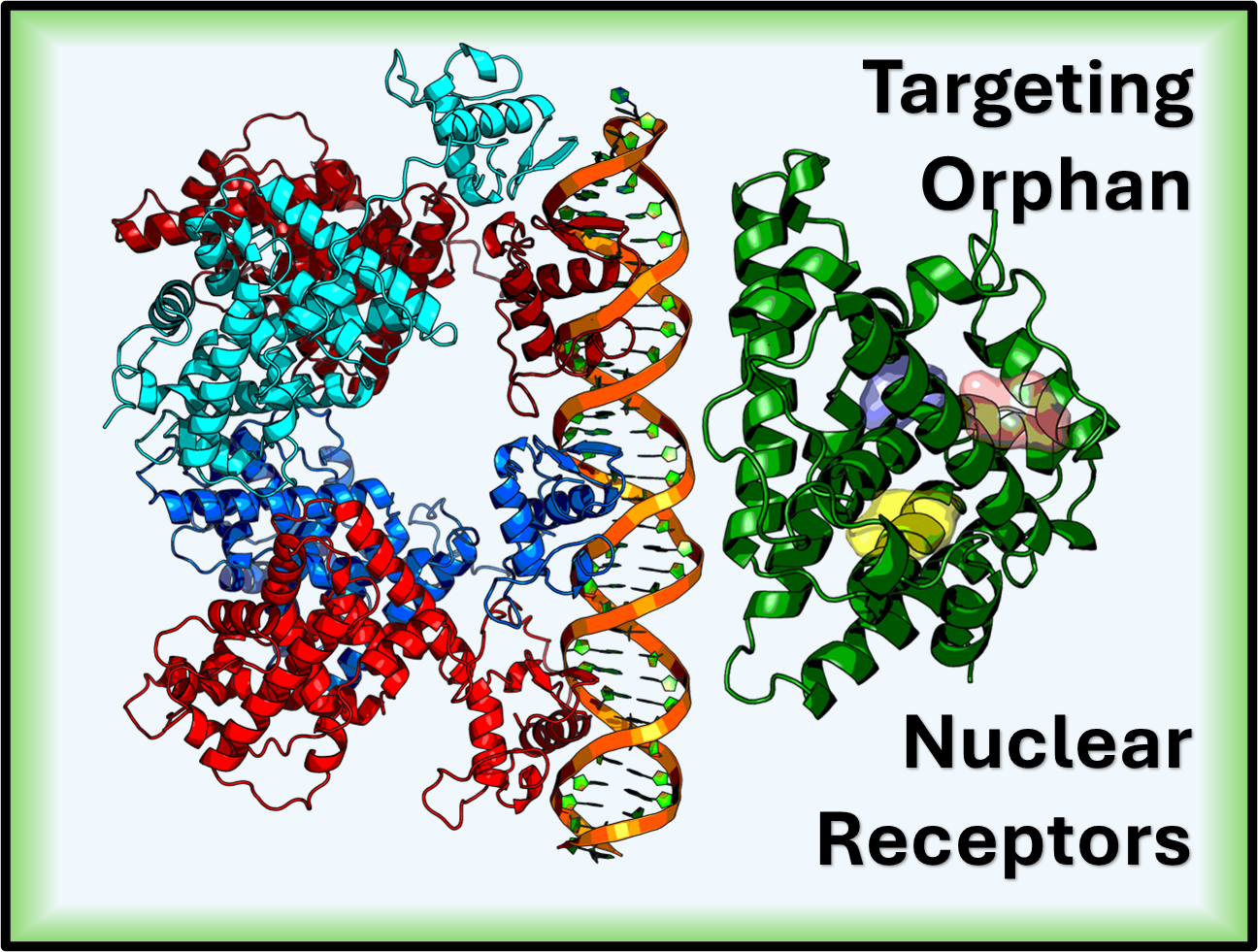
My central goal is to develop and apply next-generation computational tools to accurately predict the conformational ensembles of IDPs. These proteins, which lack a stable 3D structure, are essential for processes like gene regulation and signaling, yet are notoriously difficult to study. In this context, we are actively investigating techniques that can improve structural biology research, with a specific focus on intrinsically disordered proteins.
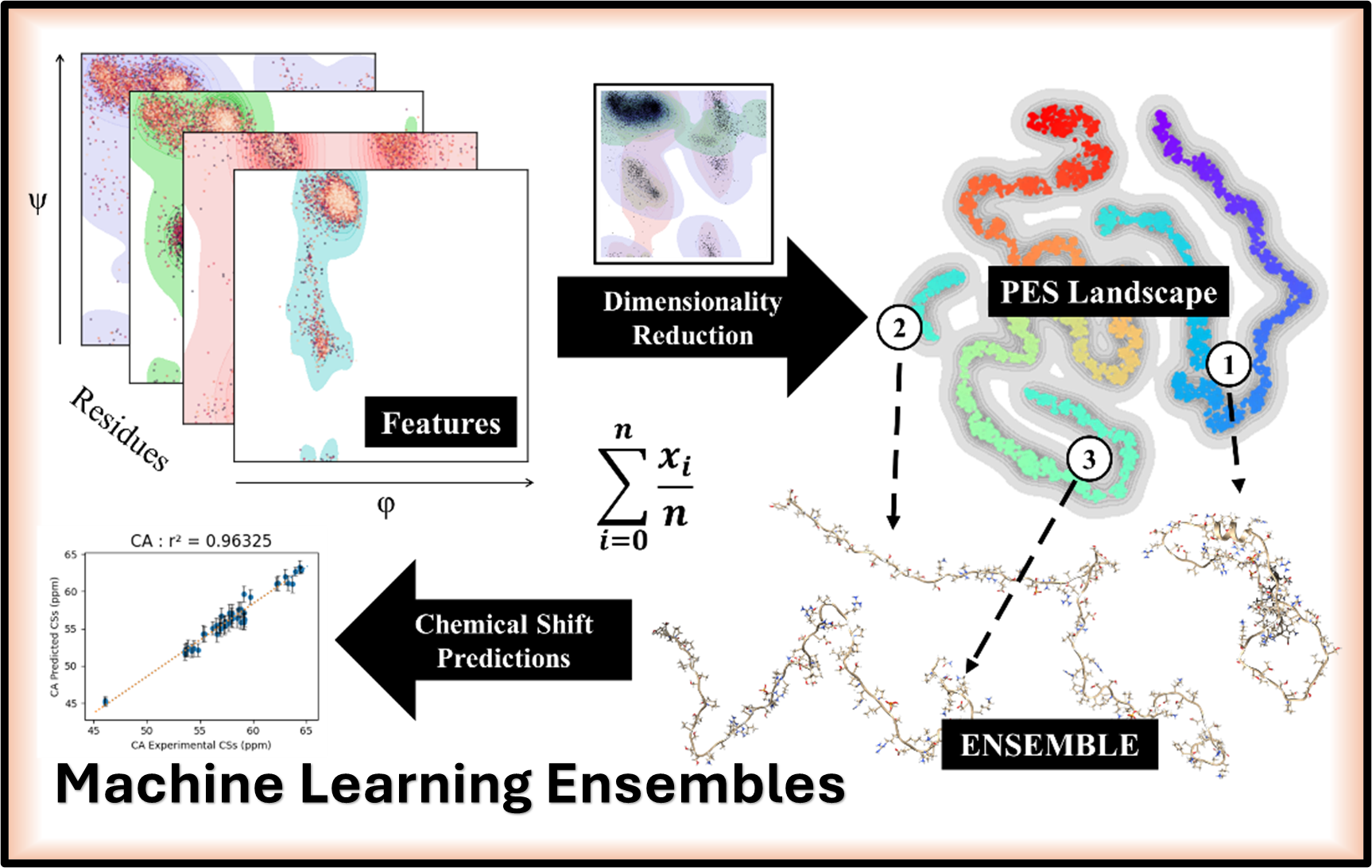
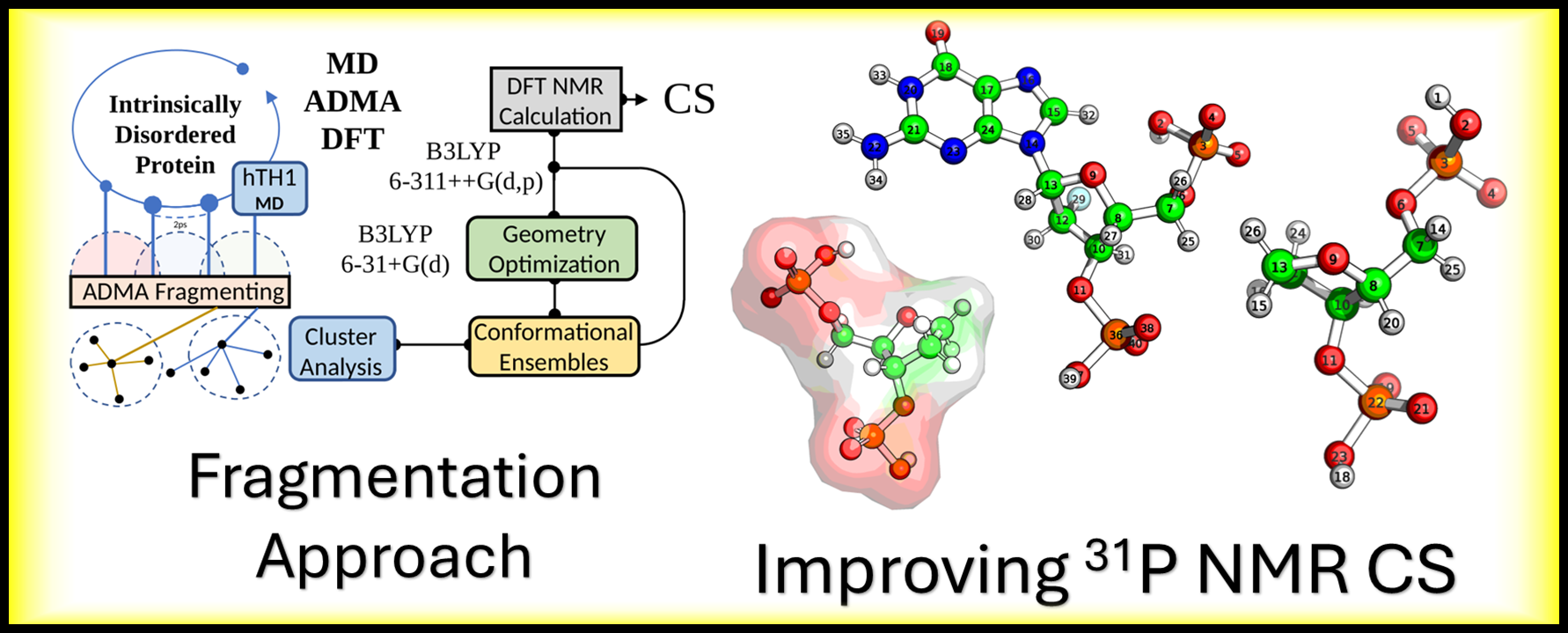
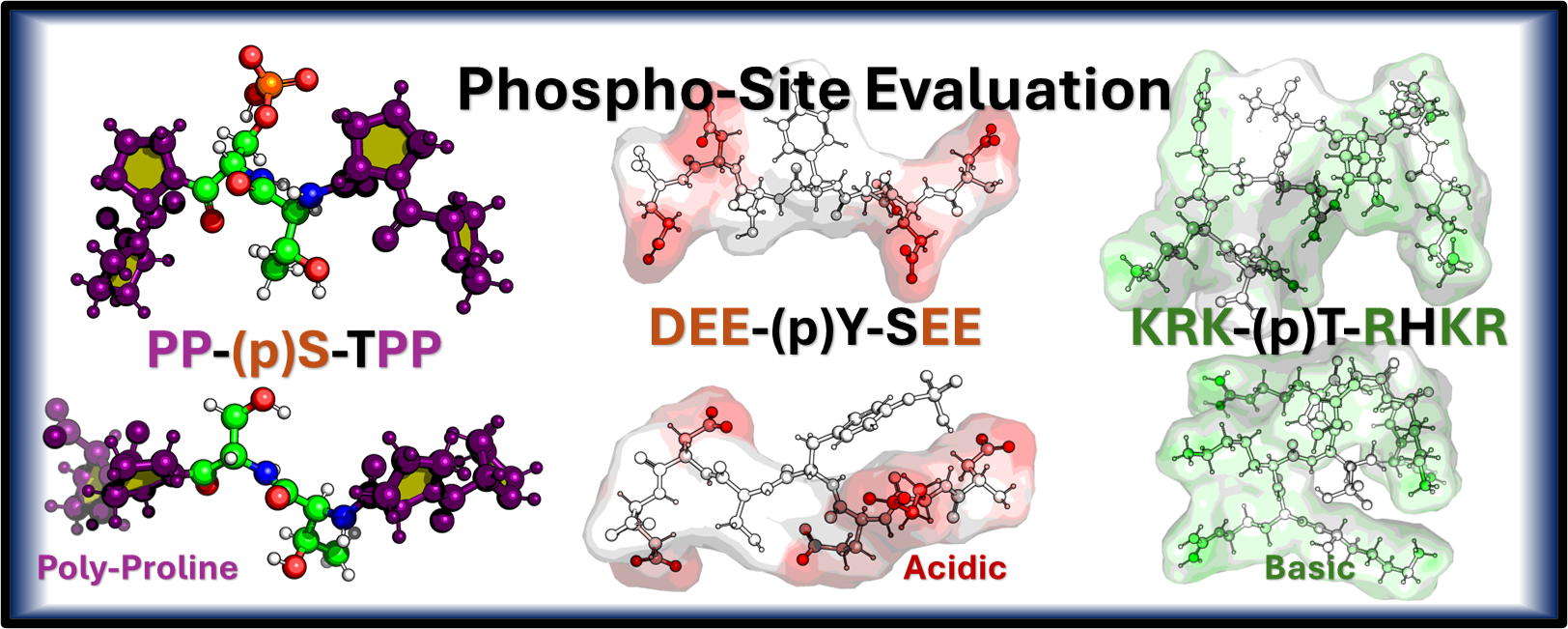
The Critical Role of p53
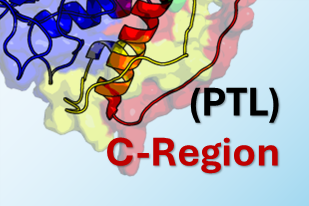
The tumor suppressor often called the “guardian of the genome” is one of the most crucial proteins in cancer biology. It functions as an that acts as a transcription factor, which either halts the cell cycle or induces apoptosis cell death in response to DNA damage. Our research is focused on this, investigating how phosphorylation, a key, affects critical Intrinsically Disordered Regions IDRs of p53, such as the pre tetramerization loop and the C terminal regulatory domain, as the flexibility of these regions is vital for the protein to form its active tetramer and correctly bind to DNA. We approached this investigation by separating the protein into four distinct computational regions, one for each of the major disordered domains of p53, namely the A-Region (TAD1/2), the B-Region (PRR), the C-Region (PTL), and the D-Region (REG). By modeling these effects using techniques like and publishing our findings, we aim to determine how PTMs stabilize or destabilize p53’s functional state, ultimately providing critical insights into cancer mechanisms and aiding in the design of therapeutics.

Beyond the Protein: Computational Applications
I also apply my expertise in computational chemistry to other critical challenges:
Sustainable Chemistry: Simulating the pyrolysis of biodiesel components (e.g., methyl linoleate) to understand reaction pathways and optimize energy conversion.
- Educational Innovation: Utilizing 3D printing technology to create physical, tactile models of molecules and proteins, enhancing complex chemistry education.
- Pharmaceutical Research: Applying statistical modeling and machine learning techniques to analyze high-throughput screening data and predict drug efficacy or toxicity. Publication in Works.