
Computational NMR Spectroscopy: Advancing Quantum Methods for Disordered Biomolecules
Project Goal: Accelerating Quantum Accuracy for Dynamic Biomolecules
This combined methodological research focuses on developing, refining, and applying advanced -based computational schemes to accurately predict and interpret chemical shifts (CSs) in complex, dynamic systems, including and . The objective is to achieve -level accuracy while ensuring and providing a detailed deconstruction of the CS signal.
Part 1: Fragment-Based NMR Prediction for IDPs

This section details the development and streamlining of a multi-scale computational framework to calculate highly accurate CSs for IDPs, focusing on efficiency and quantum accuracy.
Core Methodological Refinements (MD/ADMA/DFT)
- Efficiency through Machine Learning-Based Clustering:
- To address the high computational cost of demanding QM calculations, we implemented machine-learning based cluster analysis.
- This technique generated compact Cluster Ensembles (CLUSTER ensembles) that accurately represent the system’s dynamic behavior with significantly fewer structures than traditional sampling (REGULAR ensembles).
- Impact: A CLUSTER ensemble of just 50 structures yielded ensemble averages comparable to those obtained from a REGULAR ensemble containing 500 MD frames, dramatically reducing computational time.
- Enhanced Accuracy via Partial Geometry Optimization:
- A novel step involving partial re-optimization of the MD geometries along vibrational normal mode coordinates was introduced.
- This refinement substantially decreased errors associated with the ensemble averaging, enhancing the overall quality and stability of the computational framework.
- Optimal QM Parameters: The refined MD/ADMA/DFT framework utilized the 6-311++G(d,p) basis set in conjunction with the B3LYP functional, which was found to be the optimal choice for this application.
Part 2: Deconstructing the 31P NMR Signal in Modified Nucleic Acids
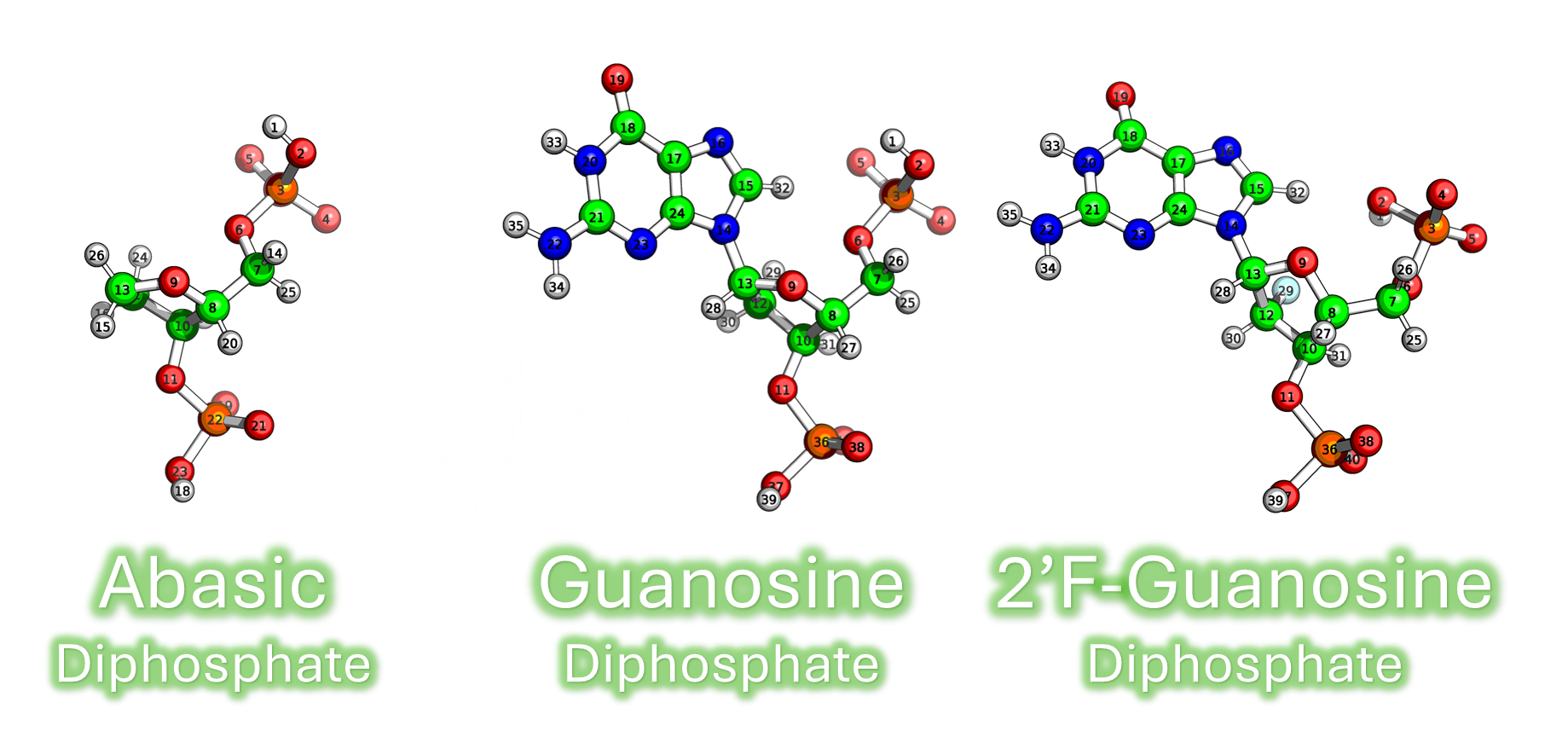
This section details the establishment and application of a Quantum Mechanics/Molecular Mechanics (QM/MM) pipeline to investigate the subtle effects of modifications in DNA/RNA on the 31P NMR CSs.
Investigation Strategy: Signal Decomposition
The project’s goal is to quantitatively separate the total experimental CS into its distinct contributions:
- Purely Electronic Effects ($\Delta\delta_{\text{Static}}$): Changes arising solely from the chemical/electronic presence of a modification (e.g., an abasic site or a $\text{}^{19}\text{F}$ label), holding the geometry constant.
- Geometric/Dynamic Effects ($\Delta\delta_{\text{Dynamic}}$): Changes caused by bond rotations and conformational flexibility.
Computational Pipeline
- QM/MM Setup: A representative nucleic acid fragment, terminated with appropriate caps and solvated with explicit ions, was established to model modified structures.
- NMR Calculation: Chemical shielding values were calculated using the GIAO method, followed by rigorous benchmarking of various functionals and basis sets.
- Decomposition: The difference between the total experimental shift ($\Delta\delta_{\text{Experimental}}$) and the purely calculated static electronic contribution ($\Delta\delta_{\text{Static}}$) provided a crucial quantitative estimation of the dynamic and geometric influence on the $^{31}$P signal.
Significance and Outlook
This combined body of work establishes state-of-the-art computational methodologies for two highly dynamic classes of biomolecules. It delivers both an efficient, high-accuracy computational engine for IDP CS prediction and a robust analytical tool for interpreting the electronic and conformational factors governing the $^{31}$P signal in modified nucleic acids, which is crucial for accurately interpreting experimental NMR data on these complex systems.
Project Collaborators

Dr. Jana Přecechtělová Pavlíková is a key collaborator, focusing on Intrinsically Disordered Proteins (IDPs), as she is a principal developer of this exact multi-scale computational methodology. Her expertise lies in coupling Molecular Dynamics (MD) with fragmentation techniques and Density Functional Theory (DFT) to accurately compute NMR chemical shifts. She has validated the use of clustering and partial re-optimization to achieve quantum-level accuracy, improving computational efficiency.

Dr. Gary Meints addresses deconstructing the 31P NMR signal in modified nucleic acids, given his extensive background in the experimental analysis of this core subject. His research group uses 31P Dynamic NMR to characterize subtle, sequence-dependent dynamics of the DNA phosphate backbone (specifically the BI/BII conformational equilibrium). This provides the crucial experimental data and physical chemistry benchmark required to validate and interpret our proposed QM/MM decomposition strategy.