
Unraveling Protein Phosphorylation Dynamics
Project Goal: Characterizing the Structural and Dynamic Consequences of Phosphorylation
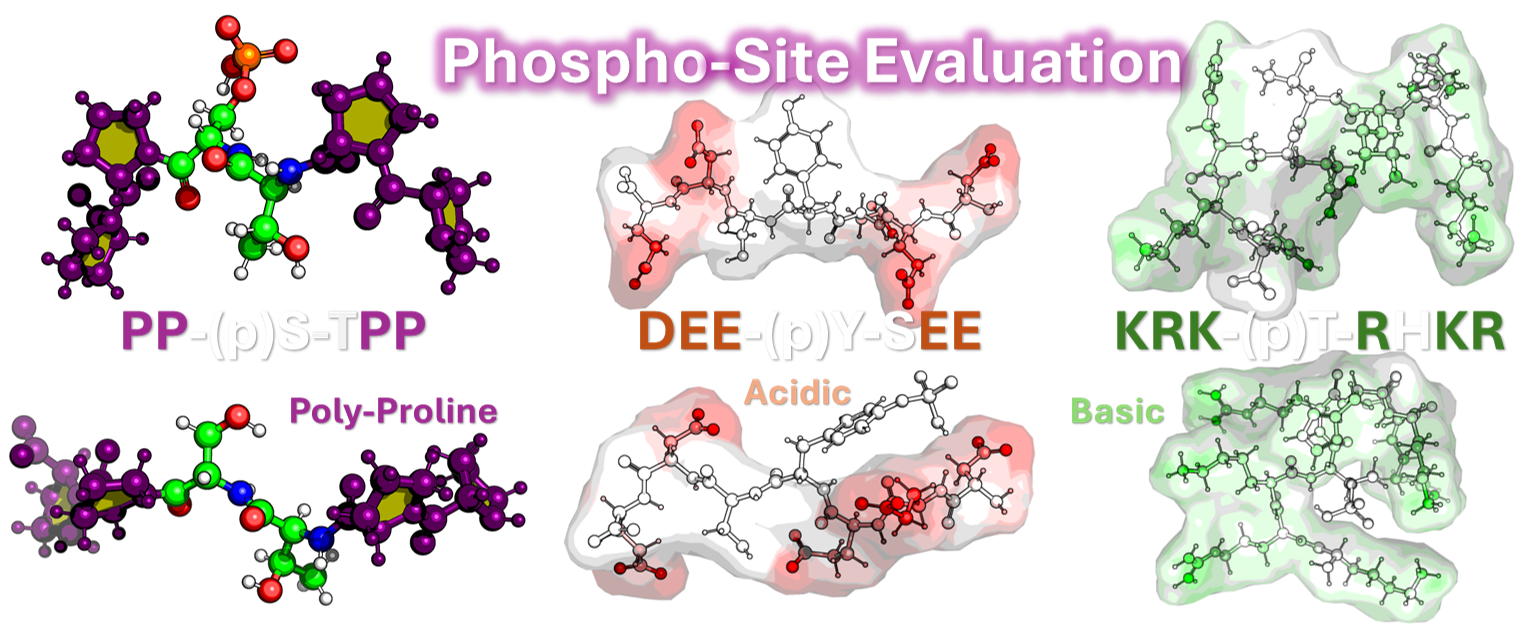
Protein phosphorylation is a fundamental regulatory mechanism crucial in numerous biological processes. The main goal of this project is to determine the structural and dynamic consequences of phosphorylation and phosphomimetic modifications on proteins and to develop computational methods for predicting these effects.
We aim to characterize how phosphorylation influences key properties, including protein size (Rg, EEdist), shape, solvent accessibility (SASA), and flexibility (RMSF). We also plan to investigate its effects on secondary structure and structural motifs, and how surrounding amino acid types (e.g., acidic, polyproline, basic regions) influence these changes.
Key Methodological Avenues
This project employs a multi-faceted approach combining computational protein design, molecular dynamics (MD) simulations, and experimental validation.
- Predictive Tool Development & Design:
- We will use machine learning techniques (scikit-learn, TensorFlow/PyTorch) to develop predictive models for phosphorylation-induced structural changes.
- We will design novel, artificial xeno-proteins that explicitly target specific motifs (like SH2 or 14,3,3 binding sites) and incorporate repeats (e.g., Proline-Rich, Glycine Repeats) to create a robust benchmark set.
- Molecular Dynamics (MD) Simulations:
- MD simulations will be employed using a combination of techniques, including all-atomistic simulations, coarse-grained simulations (MARTINI), and enhanced sampling methods (e.g., REST2).
- We will use real-world examples like Sic and pSic proteins for comparison.
- Protein Interaction & Allostery Analysis:
- The project will examine the influence of phosphorylation on protein-protein interactions, including target systems like Sic1/Cdc4, Tau/BIN1, and p27/Cyclin A-CDK2.
- We will use machine learning techniques (dimensionality reduction and clustering) to analyze MD trajectories and investigate how mutations and PTMs alter these interactions. This will involve in-silico alanine scanning combined with HADDOCK docking.
Expected Outcomes and Validation
The research is expected to produce two publications: one detailing the general structural and dynamic impact of phosphorylation and phosphomimetics, and a second focusing on the influence of phosphorylation on allostery and protein interactions in real-world systems.
Computational findings will be validated through experimental work conducted at Lund University. These experiments will utilize biophysical techniques such as Small-Angle X-ray Scattering (SAXS), Small-Angle Neutron Scattering (SANS), Circular Dichroism (CD), and Nuclear Magnetic Resonance (NMR).
Project Collaborators

Professor Marie Skepö is a leading authority on the computational biophysics of Intrinsically Disordered Proteins (IDPs) at Lund University. Her expertise is crucial for accurate Molecular Dynamics (MD) simulations and the analysis of the p53 Proline-Rich Domain (PRD). Professor Skepö rigorously links computer simulations with experimental data (e.g. CD/SAXS) to characterize complex systems. One of her focuses is the effects of phosphorylation on the stability and dynamics of disordered regions.