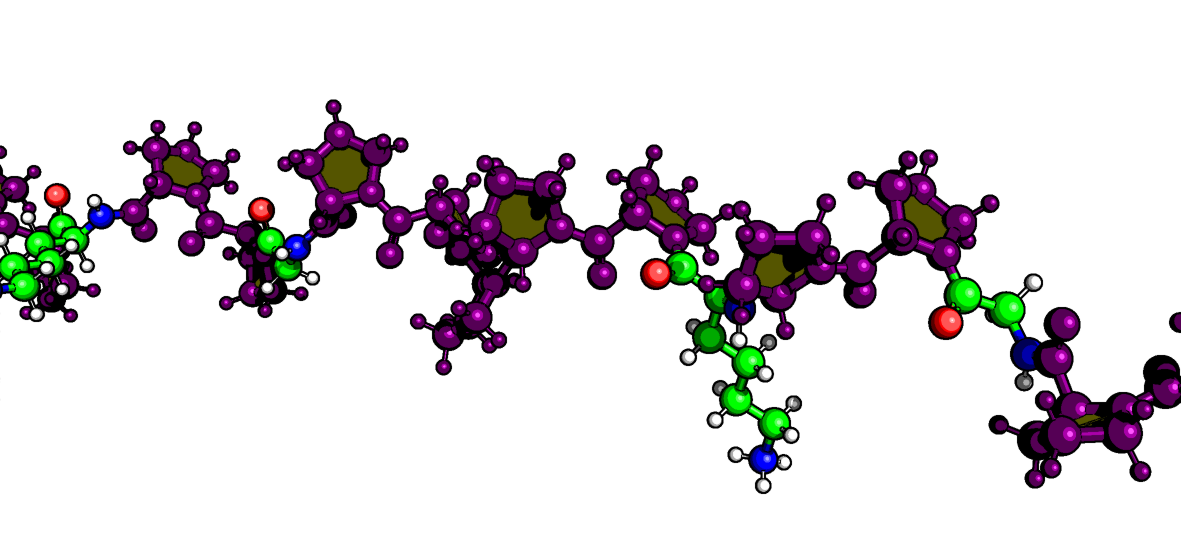
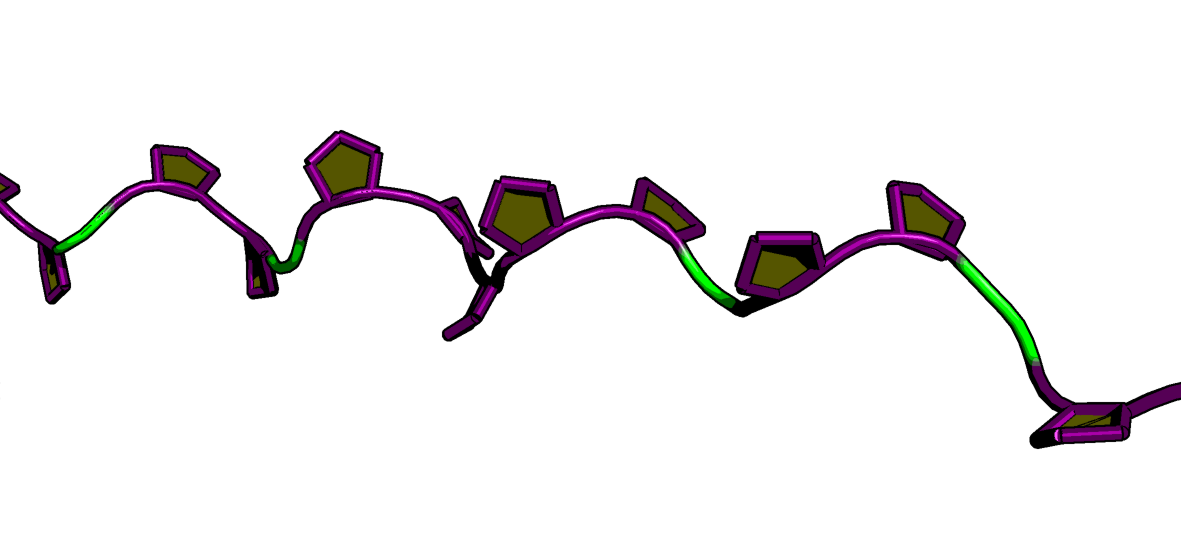
Coarse-Grained Modeling - A Quick Guide
Published:
From PDB to Production: A Hands-On Guide to Martini Coarse-Grained MD Setup
Phase 1: Environment Setup and Initial PDB Cleaning
1. Tool Installation
The necessary dependencies were installed in a dedicated cg Conda environment.
# 1. Create and activate the environment
conda create -n cg python=3.11
conda activate cg
# 2. Install core CG tools (vermouth contains martinize2)
pip install vermouth
conda install -c conda-forge mdtraj
pip install insane
# 3. Install GROMACS for simulation
conda install -c conda-forge gromacs
The original PDB file contained non-standard residues (e.g., HISE). Fix: Standardize the residue names using sed before running the conversion.
# Rename non-standard residue HISE to standard HIS
sed -i 's/HISE/HIS /g' nr2f6_dbd.pdb
This step required manually bypassing the faulty dssp executable and fixing a string-length error by supplying a contiguous 76-character Secondary Structure (SS) string.
#!/bin/bash
# FINAL 76-residue secondary structure string (H=Helix, C=Coil)
SS_STRING_76="HHHHHHHHHHCCCCCCCCHHHHHHHHHHHHHCCCCCHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCC"
martinize2 -f nr2f6_dbd.pdb \
-o nr2f6_dbd.top \
-x nr2f6_dbd_cg.pdb \
-ff martini3001 \
-p backbone \
-elastic \
-ef 500.0 -el 0.5 -eu 0.9 \
-scfix \
-cys auto \
-maxwarn 2 \
-ss "${SS_STRING_76}"
This script includes the critical step of pre-boxing the protein with gmx editconf to avoid the OverflowError bug in insane.
#!/bin/bash
INSANE_PATH="/mnt/proj2/open-33-73/miniforge3/envs/cg/bin/insane"
INPUT_CG_PDB="nr2f6_dbd_cg.pdb"
# 1. Pre-box the protein using GROMACS (Fixes fatal OverflowError in insane)
gmx editconf -f ${INPUT_CG_PDB} -o protein_boxed.gro -c -box 10.0
# 2. Solvate the system (neutralizes +11 charge with CL ions and adds 0.15M salt)
```bash
"${INSANE_PATH}" -f protein_boxed.gro \
-o system_solvated.gro \
-p nr2f6_dbd.top \
-sol W \
-box 10.0 \
-salt 0.15 \
-center
Action required: After the git clone of the Martini force fields, you must manually edit nr2f6_dbd.top:
Update File Paths: Change #include paths to the correct location within the cloned repository (e.g., martini-forcefields/.../gmx_files/martini_v3.0.0.itp).
Fix Names in [ molecules ]:
Change Protein 1 to molecule_0 1
Change CL- 11 to CL 11
Contents of em.mdp (for initial minimization):
; PARAMETER FILE FOR ENERGY MINIMIZATION
integrator = steep
emtol = 10.0
emstep = 0.01
nsteps = 50000
cutoff-scheme= Verlet
coulombtype = Reaction_Field
rvdw = 1.1
epsilon_r = 15
Contents of nvt.mdp (Constant Temperature/Volume):
define = -DPOSRES
integrator = md
dt = 0.025
nsteps = 400000
tcoupl = V-rescale
tc-grps = System
ref_t = 310
coulombtype = Reaction_Field
rvdw = 1.1
epsilon_r = 15
Contents of npt.mdp (Constant Pressure/Temperature):
define = -DPOSRES
integrator = md
dt = 0.025
nsteps = 400000
tcoupl = V-rescale
ref_t = 310
pcoupl = Berendsen
pcoupltype = Isotropic
ref_p = 1.0
coulombtype = Reaction_Field
rvdw = 1.1
epsilon_r = 15
This script addresses all known GROMACS bugs for a serial run on a small system.
#!/bin/bash
#SBATCH --job-name=Martini_EQ
#SBATCH --account=OPEN-33-73
#SBATCH --partition=qcpu
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=1
#SBATCH --time=03:00:00
#SBATCH --output=gromacs_eq.log
# Load environment and set up GROMACS
source /mnt/proj2/open-33-73/miniforge3/bin/activate cg
# STAGE 1: NVT Equilibration (Restrained Heating)
gmx grompp -c em_martini.gro -r em_martini.gro -p nr2f6_dbd.top -f nvt.mdp -o nvt_martini.tpr -maxwarn 2
gmx mdrun -deffnm nvt_martini -v -ntmpi 1
# STAGE 2: NPT Equilibration (Pressure Stabilization)
if [ -f nvt_martini.gro ]; then
gmx grompp -c nvt_martini.gro -r nvt_martini.gro -p nr2f6_dbd.top -f npt.mdp -o npt_martini.tpr -maxwarn 2
gmx mdrun -deffnm npt_martini -v -ntmpi 1
fi